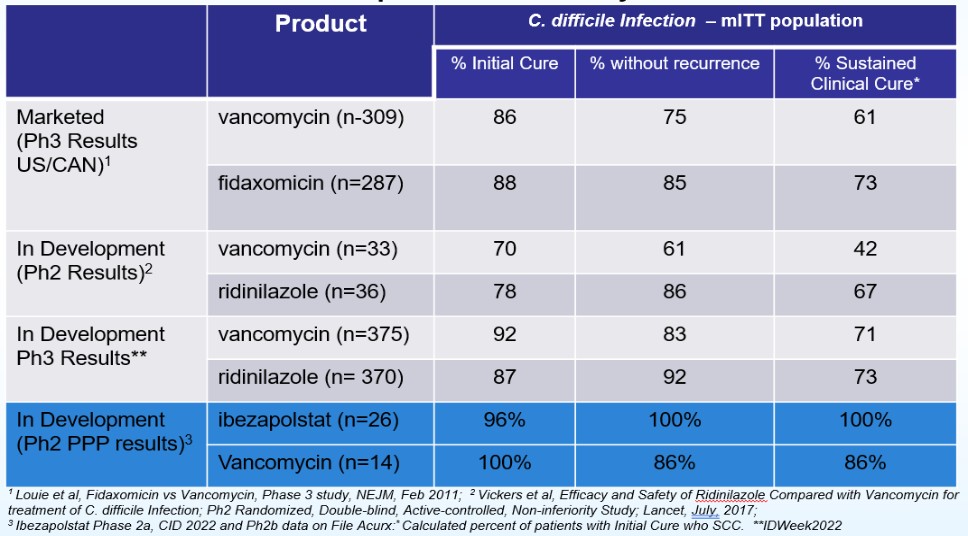
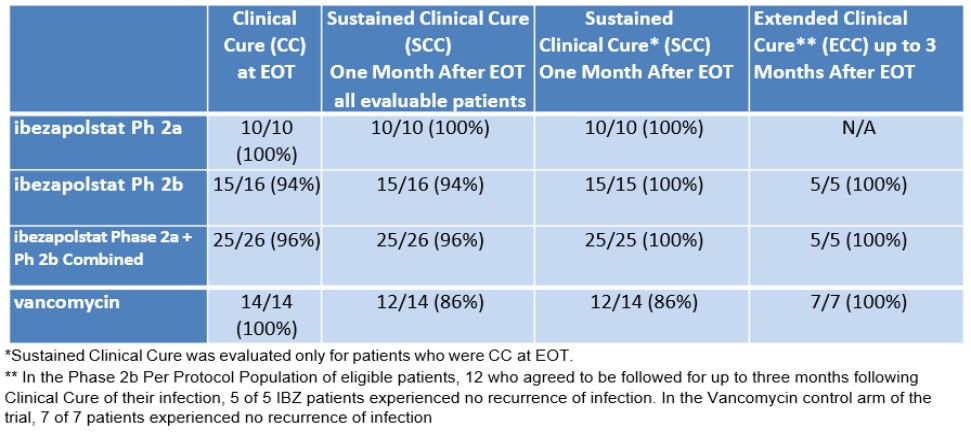
In China, there have also been recent significant developments concerning privacy and data security. The Data Security Law of the People’s Republic of China (“Data Security Law”), which took effect on September 1, 2021, requires data processing (which includes the collection, storage, use, processing, transmission, provision and publication of data), to be conducted in a legitimate and proper manner. The Data Security Law imposes data security and privacy obligations on entities and individuals carrying out data processing activities and also introduces a data classification and hierarchical protection system based on the importance of data in economic and social development and the degree of harm it may cause to national security, public interests, or legitimate rights and interests of individuals or organizations if such data are tampered with, destroyed, leaked, illegally acquired or illegally used. The appropriate level of protection measures is required to be taken for each respective category of data.
Also in China, the Personal Information Protection Law, which took effect on November 1, 2021, introduced stringent protection requirements for processing personal information, which are in many ways akin to the requirements of the GDPR. We may be required to make further significant adjustments to our business practices to comply with the personal information protection laws and regulations in China including the Personal Information Protection Law.
We also continue to see jurisdictions imposing data localization laws. These regulations may interfere with our intended business activities, inhibit our ability to expand into those markets or prohibit us from continuing to offer services in those markets without significant additional costs.
Because the interpretation and application of many privacy and data protection laws (including the GDPR), commercial frameworks, and standards are uncertain, it is possible that these laws, frameworks, and standards may be interpreted and applied in a manner that is inconsistent with our existing data management practices and policies. If so, in addition to the possibility of fines, lawsuits, breach of contract claims, and other claims and penalties, we could be required to fundamentally change our business activities and practices or modify our solutions, which could have an adverse effect on our business. Any inability to adequately address privacy and security concerns, even if unfounded, or comply with applicable privacy and security or data security laws, regulations, and policies, could result in additional cost and liability to us, damage our reputation, inhibit our ability to conduct trials, and adversely affect our business.
Our issuance of additional capital stock in connection with potential future financings, acquisitions, investments, our stock incentive plans or otherwise will dilute all other stockholders.
We expect to issue additional capital stock in the future that will result in dilution to all other stockholders. We expect to grant equity awards to employees, directors and consultants under our stock incentive plans. We may also raise capital through equity financings in the future. As part of our business strategy, we may acquire or make investments in complementary companies, products or technologies and issue equity securities to pay for any such acquisition or investment. Any such issuances of additional capital stock may cause stockholders to experience significant dilution of their ownership interests and the per share value of our common stock to decline.
Our employees, principal investigators, consultants and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and collaborators. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, to provide accurate information to the FDA and non-U.S. regulators, to comply with healthcare fraud and abuse laws and regulations in the U.S. and abroad, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices.
These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained during clinical studies that could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or