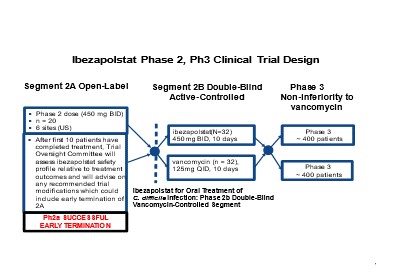
follow-up visit. No treatment-related serious adverse events (“SAEs”) were reported by the investigators who enrolled patients in the trial. We believe these results represent the first-ever clinical data showing Pol IIIC has potential as a therapeutically-relevant antibacterial target. Our Phase 2b clinical trial commenced enrollment on December 3, 2021.
The SAB is comprised of seven scientists and clinicians who have significant expertise in the scientific disciplines required for the research and development of antibiotics. The members of the SAB serve at the pleasure of management, are paid in cash on an hourly basis for their services and do not receive equity compensation. Generally, the SAB is consulted by management during the process of designing our preclinical and clinical trials as well as in the process of analyzing data generated from these trials, although the SAB’s services are not limited to such activities.
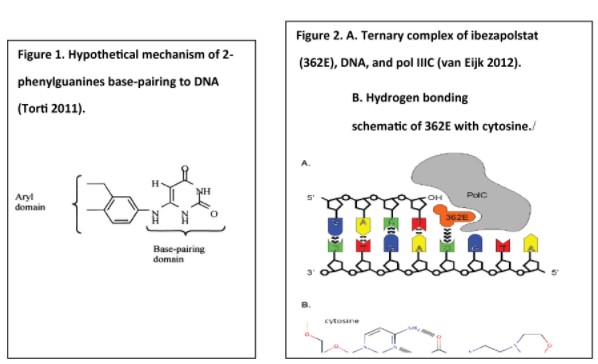
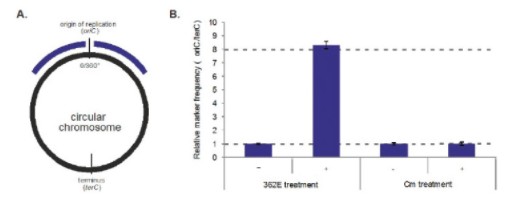
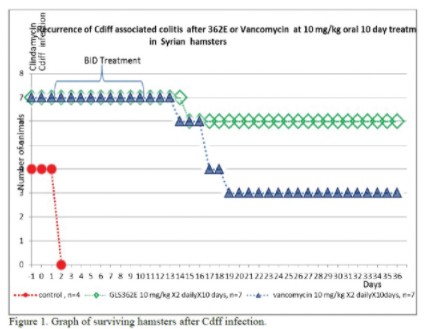
Currently available antibiotics used to treat CDI infections utilize other mechanisms of action. We believe ibezapolstat is the first antibiotic candidate to work by blocking the DNA Pol IIIC enzyme in C. difficile. This enzyme is necessary for replication of the DNA of certain Gram-positive bacteria, like C. difficile.
We also have an early-stage pipeline of antibiotic product candidates with the same previously unexploited mechanism of action which has established proof of concept in animal studies. This pipeline includes ACX-375C, a potential oral and parenteral treatment targeting Gram-positive bacteria, including MRSA, VRE and PRSP.
Our Technology
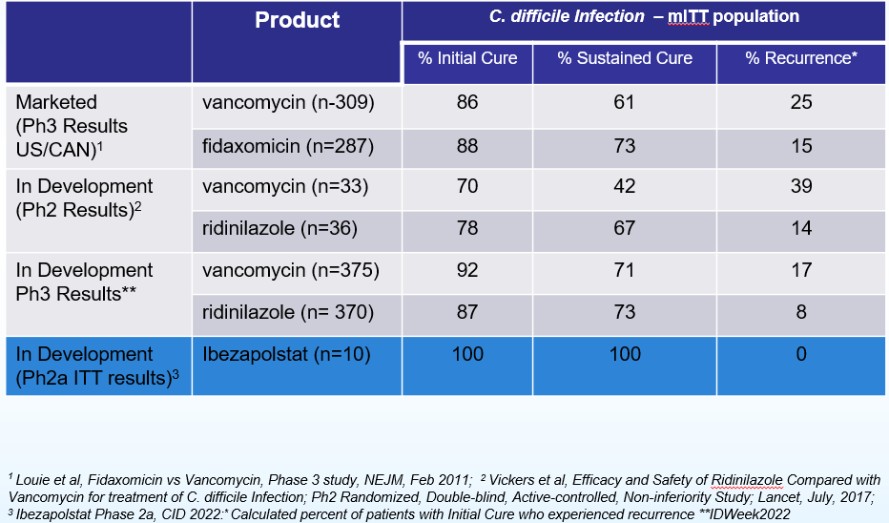
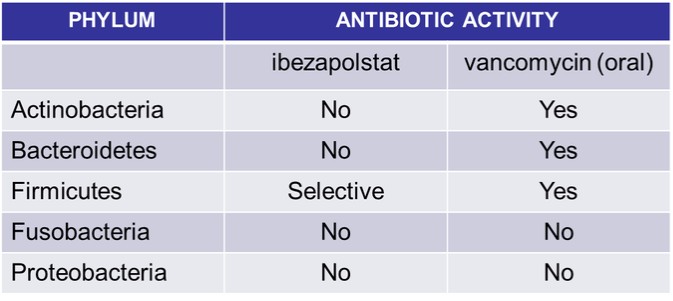
The results of our Phase 2a clinical trial also represent the first-ever clinical validation of DNA polymerase IIIC as a therapeutically relevant antibacterial target. Ibezapolstat was very well tolerated with no treatment-related SAEs noted in the Phase 2a trial. Additionally, data obtained to date demonstrate that ibezapolstat enhances actinobacteria in the microbiome and suppresses regrowth of proteobacteria; potentially lessening the likelihood of CDI recurrence or new infection by MDR Gram-negative bacteria. Additionally, the unexpected finding from further analysis of the Phase 2a study is that the beneficial Firmicutes were shown to regrow while patients were receiving ibezapolstat therapy. Several follow-up experiments have demonstrated that many of these beneficial Firmicutes have heterogeneous susceptibility to ibezapolstat allowing them to continue to perform their beneficial biologic functions even while a patient is receiving ibezapolstat for their C. difficile infection. (Garey, Oral Presentation, IDSA, IDWeek 2022 Conference, Oct 19-23, 2022).
Prior to conducting the Phase 2a clinical trial, we successfully completed a Phase 1 clinical trial of ibezapolstat for the oral treatment of CDI (the “Phase 1 Trial”). The Phase 1 Trial, conducted in the U.S., was a double-blind, placebo- controlled study to determine safety, tolerability, pharmacokinetics (“PK”) and fecal concentrations of ibezapolstat in 62 healthy volunteers. It was conducted in two parts; first, single ascending doses were administered to four cohorts of eight subjects each, and second, multiple ascending doses were administered that simulate the anticipated clinical treatment regimen. Safety information was analyzed through assessment of adverse events and other standard safety measures, while concentrations of ibezapolstat were determined in both blood and the feces, the latter being the critical site of drug delivery for treating CDI. In addition, the laboratory of Dr. Kevin Garey at the University of Houston performed state-
4